Researchers at CNCR-FGA and two Italian institutes describe the first two patients with a homozygous STXBP1 mutation. Opposite to known mutations, this mutation increases synaptic transmission. The study is published in the leading journal Brain.
STXBP1 mutations cause a neurodevelopmental disorder, STXBP1 syndrome, characterized by a developmental delay, intellectual disability, and in the majority of cases epileptic seizures and movement disorders. Research into the disease mechanisms underlying STXBP1 syndrome is a main focus of CNCR-FGA.
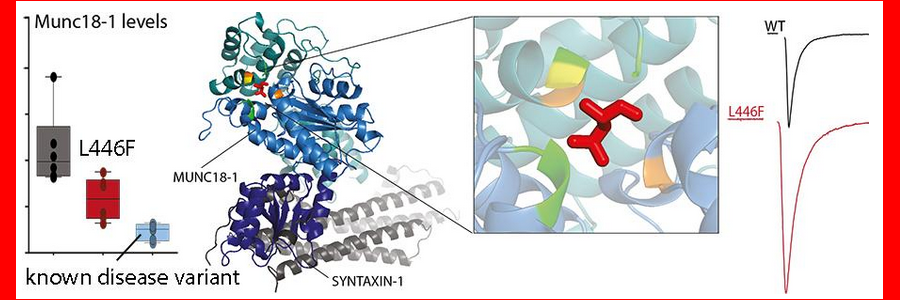
All symptom-causing STXBP1 mutations reported to date are heterozygous. However, in this study, a novel mutation, L446F, is identified in two homozygous carriers, by physicians and geneticists at the Gaslini Institute in Genoa and University Magna Graecia in Catanzaro. Both patients show the main hallmarks of STXBP1 syndrome, whereas heterozygous carriers of the same variant are asymptomatic.
Annemiek van Berkel and Hanna Lammertse (FGA) have modelled the mutation in rodent neurons to investigate the cellular consequences of this unique mutation. They show that the stability of the STXBP1 gene product MUNC18-1 is affected, but to a lesser extent than for known heterozygous disease mutations. Moreover, the effect of the L446F mutation on synaptic transmission is the opposite of what is observed for heterozygous disease mutations: neurons expressing the L446F mutation show a gain-of-function in synaptic transmission, as opposed to a loss-of-function observed previously for heterozygous disease mutations (Kovacevic et al, 2018).
These findings suggest that mutations that have a very different effect at the level of single neurons, may eventually lead to a similar net effect at the level of brain activity patterns.
The paper was published on December 19th 2019 and is available here.