Research

The department of Functional Genomics (FGA) headed by Professor Matthijs Verhage has over 20 years of experience and published more than 40 peer reviewed papers dissecting the cellular function(s) of STXBP1/MUNC18-1 in the brain. FGA is part of the Center for Neurogenomics and Cognitive Research based at the Vrije Universiteit and the Amsterdam UMC.
The following information serves to give insight into the functions of MUNC18-1 that were discovered by our lab in the last two decades. We have identified STXBP1 as a central factor in synaptic transmission and provided insight into the molecular and intramolecular mechanisms of STXBP1 function. Furthermore, we have mapped several cellular pathways that regulate MUNC18-1 function, especially phosphorylation by PKC, ERK and neuronal Src family kinases. Initially, these studies were performed using mouse (cellular) models. Currently, we are expanding our model systems from rodent to human neurons, allowing the analysis of the effect of STXBP1 gene defects in a human (patient) genetic background.
Fundamental research on Munc18-1
Munc18-1 is a key player in neurotransmission


In 2000, we discovered that, in mice, in the absence of Munc18-1, synaptic vesicles cannot fuse. While neurotransmitter-filled vesicles are still located at the synapse, no fusion events are observed, neither spontaneous events nor responses to action potential stimulation, chemical depolarization or other agents that normally trigger fusion. This defect is absolute (not a single event is ever observed) and has been observed in different synapses, from the CNS to the neuromuscular junction (Verhage et al., 2000). These findings showed that Munc18-1 is essential for neurotransmission.
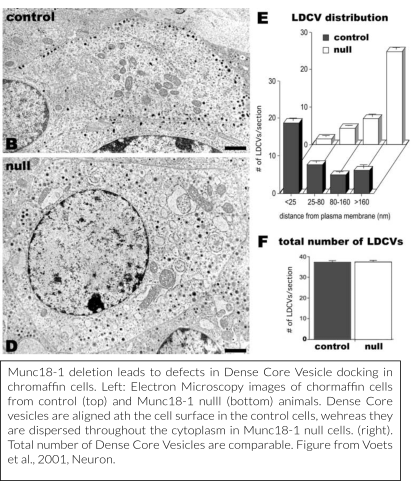
In subsequent studies, the synaptic role of Munc18-1 was further characterized. To this end, we have generated knockout (Munc18-1 null) or conditional knockout (Munc18-1 lox) mouse lines. From these, primary (post-mitotic) neurons and adrenal chromaffin cells were used for experimental analysis.
In the knockout background, mutant versions of Munc18-1 were introduced to address the role of specific amino acid residues, protein modifications, such as phosphorylation, or protein domains of the protein.
Together with research teams in Göttingen, Germany (the lab of prof. Neher, dr. Klingauf and dr. Moser) and Copenhagen, Denmark (prof. Sørensen), we have provided detailed models of how Munc18-1 regulates secretory vesicle docking (Voets et al., 2001, Toonen et al., 2006, De Wit et al., 2009) and subsequently characterized, also in collaboration with a research team in Heidelberg (prof. Söllner), downstream functions in vesicle priming and in preventing de-priming (Gulyás-Kovács et al., 2007, Munch et al., 2016, He et al., 2017). Our most recent mechanistic work investigates the sub-molecular configurations of the protein that help vesicles to transit from the initial docking to the primed state to fusion by accommodating sequential interactions with Syntaxin-1, SNAP25 and Synaptobrevin/VAMP2 (Munch et al., 2016, Meijer et al., 2017), without the necessity for the connection after SNARE-complexes have formed (Meijer et al., 2012).
We have shown that the two known splice variants of Munc18-1 have very similar roles in the synapse, but modulate presynaptic short-term plasticity in a slightly differential manner (Meijer et al., 2015).
Protein expression levels influence function
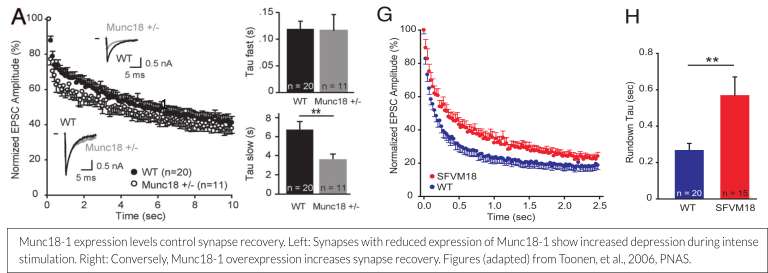
The amount of Munc18-1 expressed at the synapse or in a chromaffin cell is a determinant for the size of the readily releasable vesicle pool (RRP). Munc18-1 overexpression leads to a 50% increase in pool size and a concomitant delayed run down during repetitive stimulation, whereas a 50% reduction of Munc18-1 levels leads to early run down (see figure below; Toonen et al., 2006, Wierda et al., 2007). These findings are increasingly relevant, as the disease mechanism for STXBP1 Encephalopathy is currently believed to be haploinsufficiency (more information on this can be found here – scroll down to ‘Haploinsufficiency: A likely disease mechanism’).
Munc18-1’s main interaction partner is Syntaxin-1 and most of its known functions work in concert with this protein. Moreover, in the absence of Munc18-1, also Syntaxin-1 levels are reduced by 70% (Toonen et al., 2005). This dependence between Munc18-1 and Syntaxin-1 is reciprocal, as Munc18-1 levels are also reduced upon Syntaxin-1 deficiency. This interdependency is not observed with other synaptic proteins (Gerber et al., 2008, Vardar et al., 2016).
Munc18-1 is a key element for adaptation and modulation of neurotransmission
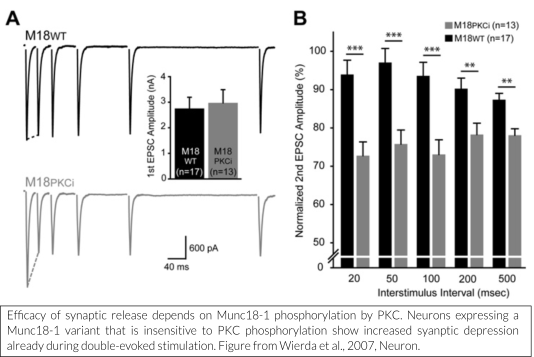
Some of the major mechanisms to modulate neurotransmission target Munc18-1 as one of the main players for modulation.
One such mechanism is the diacylglycerol (DAG)/protein kinase C (PKC) pathway, known to enhance neuronal transmission. We have shown that Munc18-1 is rapidly phosphorylated by PKC in response to strong stimulation (De Vries et al., 2000). This leads to phosphorylation-dependent translocation of Munc18-1 (Cijsouw et al., 2014).  In addition, PKC-dependent phosphorylation of Munc18-1 is required for DAG-dependent enhancement of neuronal transmission, probably in conjunction with activation of Munc13 (Wierda et al., 2007, Genc et al., 2014).
In addition, PKC-dependent phosphorylation of Munc18-1 is required for DAG-dependent enhancement of neuronal transmission, probably in conjunction with activation of Munc13 (Wierda et al., 2007, Genc et al., 2014).
Other modulatory mechanisms can act via the activation of presynaptic metabotropic glutamate receptors (mGluR) or of cannabinoid receptors (CB1R), which leads to inhibition of neurotransmission. We have shown that ERK-dependent phosphorylation of Munc18-1 is an essential downstream event in mGluR and CB1R activated synaptic inhibition (Schmitz et al., 2016).
Finally, we have shown that tyrosine phosphorylation of Munc18-1 by Src-kinases and possibly other (unknown) kinases is a powerful way to inhibit synaptic transmission (Meijer et al., 2017).
Munc18-1 is involved in many regulated secretory pathways
The evidence for the role of Munc18-1 in neurotransmission and regulated secretion in chromaffin cells is robust and undisputed (for reviews see Toonen and Verhage., 2003, Toonen and Verhage., 2007). In addition, we have shown that Munc18-1 is required for regulated secretion from neurosecretory terminals in the anterior pituitary, resulting in a dramatic reduction in the levels of circulating pituitary hormones (Korteweg et al., 2005). The expression of neuropeptide genes is reduced in the neocortex of Munc18-1 null mice, as revealed by microarray analysis and qPCR (Bouwman et al., 2006). In collaboration with a research team in the US (prof. Thurmond) we have shown that Munc18-1 regulates the 1st phase of insulin secretion in the pancreas (Oh et al., 2012). Hence, we conclude that Munc18-1 is important for most regulated secretory pathways in our body. Munc18-3 (also called Munc18c) supports regulated exocytosis of glucose transporter vesicles in muscle and fat cells (Thurmond et al., 1998). 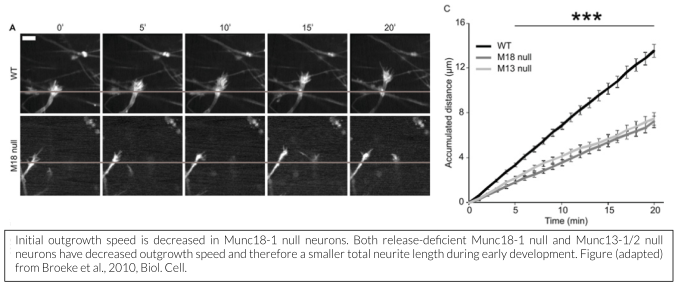
Munc18-1 plays a role in synaptic development
While membrane incorporation via exocytosis is generally considered to contribute to neurite outgrowth, we showed that Munc18-1 is not essential for outgrowth and initial synapse development, but that filopodia of outgrowing growth cones are hypermotile and the base of the cone progresses slower in the absence of Munc18-1 (Broeke et al., 2010). Munc18-1 does play a crucial role in synapse maintenance (Heeroma et al., 2003, Bouwman et al., 2004, Korteweg et al., 2004), but this may be an indirect effect, 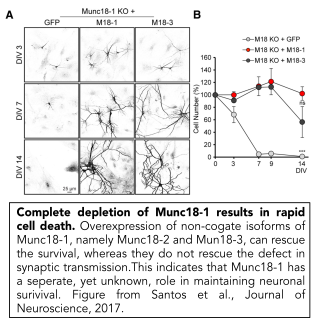 related to the role of Munc18-1 in cell viability (see next section).
related to the role of Munc18-1 in cell viability (see next section).
A separate role for Munc18-1 in cell viability
In addition to its well-described role in synaptic transmission, we have discovered that Munc18-1 plays a crucial role in neuronal survival (Verhage et al., 2000, Heeroma et al., 2004, Santos et al., 2017). Neurons that cannot express Munc18-1 die in a cell-intrinsic manner. Their survival can be extended, but not rescued by trophic factors such as insulin or BDNF (Heeroma et al., 2004). We showed that neuronal survival in Munc18-1 null neurons is rescued by over-expressing other, non-cognate Munc18 genes, Munc18-2 and Munc18-3, that are normally expressed only at low levels in the brain. Expression of either of these genes rescues neuronal viability, but not synaptic transmission (Santos et al., 2017). This indicates that Munc18-1 has an essential, yet unknown, role in neuronal viability which is unrelated to synaptic transmission. Ongoing research in the lab is dedicated to find this essential function.
Translational & clinical research
In addition to our research on the fundamental role of STXBP1/MUNC18-1 in neurons, we also dedicate our work to the understanding of how mutations in STXBP1 lead to STXBP1 Encephalopathy. On our page for patients, as well as for clinicians, you can find more information on STXBP1-E. Here, we briefly outline our current research lines.
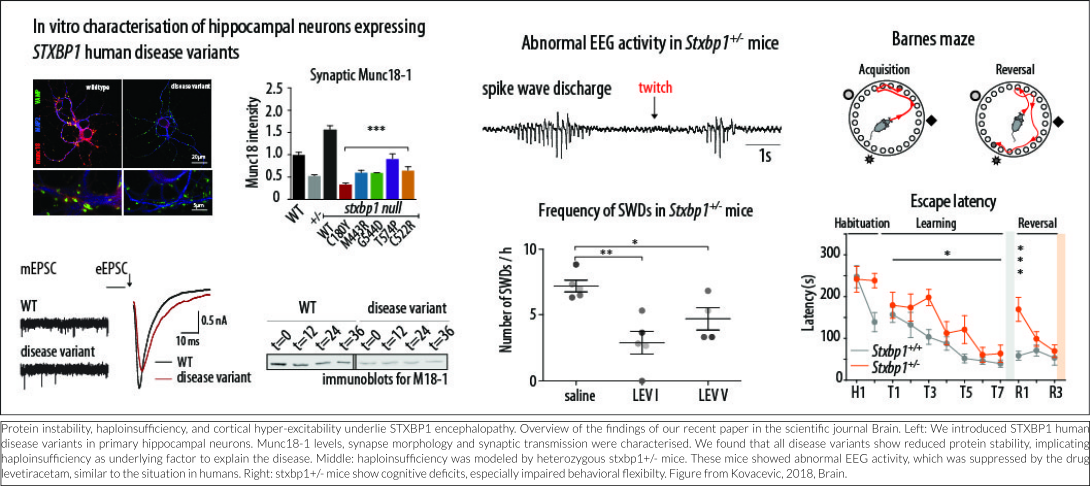
Heterozygous deletion of STXBP1 in mice recapitulates symptoms shown in STXBP1-E
No correlation is apparent between the predicted detrimental effect of mutations in STXBP1 and symptom severity. One explanation for this is that all disease-causing mutations lead to reduced protein stability, resulting in haploinsufficiency (Stamberger et al., 2016). Several lines of evidence from our lab support this hypothesis, which were recently published in the peer-reviewed journal Brain (Kovacevic et al., 2018). In this study, in silico analyses of publically available genomics data of hundreds of thousands of humans show that pathogenic mutations in the STXBP1 gene are ultra-rare, probably due to extremely strong consequences of STXBP1 mutations for normal brain development and function. Expression of mutated STXBP1 proteins in a cell culture system (in vitro) show that all mutations lead to a decreased cellular expression level of the STXBP1 protein, implicating protein instability as the underlying factor to explain the disease. STXBP1 heterozygous (STXBP1+/-) mice have one functional copy of the gene and therefore provide a valid animal model for STXBP1 haploinsufficiency. Three lines of STXBP1+/- mice were characterized, on different genetic background or by deleting expression in GABA-ergic neurons only, and report spontaneous epilepsy-like behavior and abnormal EEG activity, remarkably similar to human patients. Interestingly, excessive neural activity was detected not in the brain regions most typically involved in seizure activity, but in the more superficial neocortical regions of the brain. This suggests that increased cortical excitability is the underlying cause of disease at least in STXBP1+/- mice. New EEG analysis in STXBP1 Encephalopathy patients will elucidate whether this neocortical hyper-excitability is indeed an underlying cause in patients. In STXBP1+/- mice, the abnormal EEG activity was suppressed by the antiepileptic drug levetiracetam, again similar to the situation in humans. In addition, STXBP1+/- mice showed cognitive deficits, especially impaired behavioral flexibility: an impaired ability to suppress previously learned responses and to learn new. Taken together, these mouse models are valuable tools for future therapy design because they model the situation in patients closely (construct validity), they show strikingly similar deficits (face validity) and respond in the same way to the same intervention (predictive validity).
Current research lines on STXBP1 Encephalopathy
Our long standing research on STXBP1 and MUNC18-1 has sparked new ideas on how STXBP1 mutations cause the symptoms that are experienced by the patient. Currently, we are exploring these ideas in three research lines:
Investigating the relationship between mutations and symptoms
Background
Mutations in STXBP1 typically cause changes in the structure of the STXBP1 protein. Based on similarities and dissimilarities between the normal and mutated protein, we can predict how strong a given mutation will affect the function of the STXBP1 protein inside our cells. Strikingly, for STXBP-E patients studied so far, no correlation was observed between the predicted severity of mutations and the severity of the symptoms. In other words, if a mutation in STXBP1 is predicted to have a strong effect on protein function, the symptoms are not more severe than for a mutation with predicted mild effect.
Our aim
Because it might be that certain characteristic features or symptoms experienced by multiple patients are currently overlooked because information tends to be gathered from multiple sources (such as separate clinical reports), we think it is important to study these relationships between mutations and symptoms more systematically in a large group of STXBP1-E patients. Therefore, our goal is to obtain a more complete and standardized picture of all symptoms for each individual patient, with each specific mutation, and make better comparisons between symptoms and mutations. This can help in reaching a faster diagnosis for new STXBP1-E patients. Moreover, it could help physicians to make better prognoses on the occurrence of particular symptoms for their patients, based on their mutation.
Performing non-invasive EEG recordings to analyze excitation/inhibition balance
Background
There are two dominant types of synapses: excitatory synapses, which activate other nerve cells, and inhibitory synapses, which inhibit other nerve cells. For optimal brain function, a delicate balance between excitation and inhibition is maintained. Too much excitation can lead to epileptic activity, whereas too much inhibition can suppress brain activity. This delicate balance is called the excitation/inhibition (E/I) balance. Our work in mouse models for STXBP1 Encephalopathy shows that the loss of functional STXBP1 may affect the E/I balance by affecting the performance of inhibitory synapses more than excitatory synapses. This might imply that there is reduced inhibition in patients carrying STXBP1 mutations, thereby shifting the E/I balance towards over-excitation, leading to epileptic seizures and disturbances in cognitive performance.
Our aim
We aim to test this idea by measuring brain activity in STXBP1-E patients and compare this to recordings from individuals without an STXBP1 mutation. Brain activity will be measured using electroencephalography (EEG) recordings. Understanding whether an imbalance between excitation and inhibition occurs in the brain of a STXBP1-E patient could give valuable information on mechanisms that may be underlying the symptoms experienced by the patient. In the future, this could also be used to screen for, and develop, new therapeutic strategies.
Studying the effects of STXBP1 mutations in single and small networks of nerve cells
Background
Haploinsufficiency is the most likely explanation for STXBP1-E. Nevertheless, this cannot explain the wide range in observed symptoms between patients. Possibly, other genetic factors have an additional effect. An important step we have recently taken is to gather cellular material from patients via skin biopsies (on so-called clinic days, see here). We can re-program the skin cells obtained from these patients to a stem cell state (induced pluripotent stem cells, iPSCs) and subsequently differentiate these to neurons in a culture dish. This provides us with the unique opportunity to study the pathophysiology of STXBP1 mutations in vitro using neurons with the exact genetic background of STXBP1-E patients.
Our aims
Using patient-derived cells, we may be able to identify cellular processes that are altered as a consequence of a 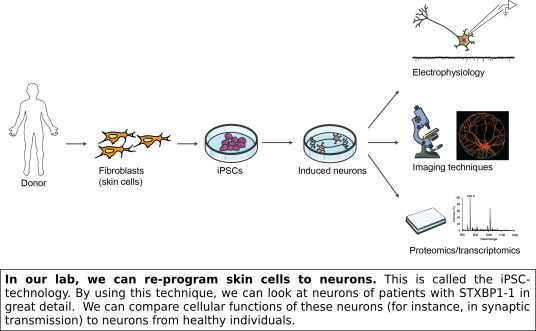 mutation in STXBP1. Knowing which cellular processes are affected will allow us to gain a more thorough understanding of pathological mechanisms underlying the symptoms that patients suffer from, and in turn may lead to new hypotheses about ways to help restore these cellular processes. This cell model could in the future be used to screen new treatment options.
mutation in STXBP1. Knowing which cellular processes are affected will allow us to gain a more thorough understanding of pathological mechanisms underlying the symptoms that patients suffer from, and in turn may lead to new hypotheses about ways to help restore these cellular processes. This cell model could in the future be used to screen new treatment options.